
El síndrome de Prader-Willi (SPW) es consecuencia de una alteración genética originada por la falla en la expresión de genes del cromosoma 15.
En la etapa de lactancia se caracteriza por hipotonía y dificultad para succionar, lo que ocasiona un retraso en el crecimiento. Posteriormente, durante la infancia, se produce un retraso en el desarrollo psicomotor junto con discapacidad intelectual y problemas en el comportamiento. La enfermedad cursa con una deficiencia en la producción de hormonas del eje hipotalámico-hipofisario-adrenal, del crecimiento, gonadotrofinas y tiroideas, ocasionando obesidad, apetito excesivo, tendencia a padecer diabetes,alteraciones en el control de la temperatura, capacidad baja de sentir dolor, trastornos de la respiración al dormir, alteraciones del sueño, junto con otros problemas.
HISTORIA
Fue descrita en el año 1956 por los doctores suizos Andrea Prader, Alexis Labhart y Heinrich Willi, en nueve pacientes que presentaban un cuadro clínico de obesidad, talla baja, hipogonadismo, criptorquidia y alteraciones en el aprendizaje tras una etapa de hipotonía muscular pre- y posnatal, además de una discapacidad intelectual de leve a moderada.
EPIDEMIOLOGÍA
La incidencia y frecuencia publicada es muy variable, aceptándose que entre 1 de cada 10.000 niños y 1 de cada 30.000 niñas (en función de las poblaciones) nace con esta compleja alteración genética. Considerada una enfermedad rara, parte de su complejidad se basa en el amplio rango de manifestaciones clínicas y en su variable grado de severidad.
ETIOLOGÍA
El síndrome de Prader-Willi es una enfermedad genética producida por la ausencia de la expresión de un alelo localizado en el brazo largo del cromosoma 15 de origen paterno (concretamente en la región 15q11-q13), el alelo materno no se expresa debido al fenómeno de la impronta genética. Esta ausencia de expresión puede deberse a varias causas, y es por ello que la herencia de este síndrome es compleja.
En individuos sanos, el cromosoma paterno expresa varios genes (SNRPN, NDN, MAGEL2, cluster snARN), e inhibe la del gen UBE3A. Lo hace porque no se encuentra metilada la diana de impronta génica. En cambio, el cromosoma materno sí tiene metilada esta diana, por lo que inhibe la expresión de los genes que se expresaban en el paterno y activa la de UBE3A. El efecto fundamental es que se deja de sintetizar una ribonucleoproteína. Esto es lo que ocurre en condiciones normales, en las que los óvulos y espermatozoides realizan el proceso de impronta génica correctamente. El síndrome de Prader-Willi se produce al faltar la expresión de los genes que un cromosoma paterno silvestre o sano expresaría. La falta de esta expresión puede ser debida a varias causas:
- Deleción o pérdida de la región 15q11-q13 del cromosoma de origen paterno. Se observa en el 70% de los pacientes. El riesgo de recurrencia familiar es cercano al 1%
- Disomía uniparental materna, es decir, la presencia de dos cromosomas maternos en vez de uno paterno y otro materno. Se observa en aproximadamente un 20% de los pacientes. El riesgo de recurrencia no supera el 1%
- Defectos en la impronta de la diana que silencien los genes que deberían expresarse en el paterno. En este caso aunque la frecuencia es menor, si se presenta el riesgo de recurrencia puede llegar al 50%
Es importante llamar aquí la atención sobre la semejanza génica de este síndrome con el de Angelman, en el que la enfermedad se desarrolla por la ausencia de expresión de varios alelos en el mismo locus, pero en este caso, de origen materno.
CUADRO CLÍNICO
Este síndrome altera el funcionamiento del hipotálamo, una sección del diencéfalo cuyas funciones incluyen, entre otras, el control del apetito lo que provoca que carezcan de sensación de saciedad. Debe distinguirse entre sensación de hambre y falta de saciedad. Un error muy común es pensar que la búsqueda incesante de comida” se debe a un hambre excesiva. La alimentación de las personas con SPW necesita estar supervisada constantemente, además de seguir una dieta estricta para evitar la obesidad.
Provoca asimismo deficiencia del tono muscular, un alto porcentaje de grasa en el organismo y falta de energía. Todas estas condiciones reducen las necesidades calóricas de los niños y adultos que tienen este síndrome a dos tercios de la necesidad calórica estándar.
Si bien el trastorno alimenticio es el síntoma más evidente y el que demanda más tiempo, además de su mayor riesgo vital, es sólo un aspecto de esta compleja dolencia. Al principio, los bebés que tienen este síndrome se alimentan de forma deficiente y no aumentan de peso, ya que la debilidad de su tono muscular reduce su capacidad de succión.
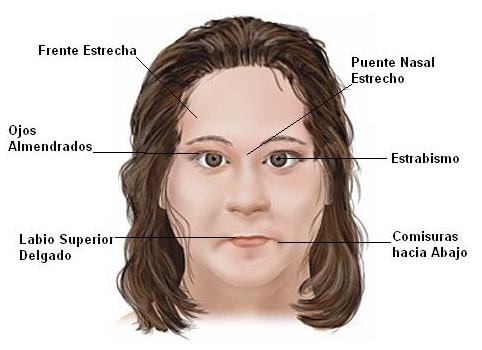
El síndrome de Prader-Willi también puede provocar crecimiento y maduración incompletos, facciones características, problemas del comportamiento, dificultades respiratorias, comportamiento obsesivo-compulsivo (como hurgarse en lesiones en la piel, pensamientos y acciones repetitivos y una fuerte necesidad de seguir una rutina), disfunciones en la temperatura corporal, resistencia al dolor, retraso en el desarrollo del aprendizaje y, en dos terceras partes de los casos, imposibilidad de vomitar. La hipopigmentación suele ser también una característica de estos pacientes.
DIAGNOSTICO Y TRATAMIENTO
Aunque no existe tratamiento para curar la enfermedad, el diagnóstico temprano es muy importante. Las razones básicas son anticiparse a las complicaciones que puede presentar la persona y ofrecer un consejo genético, por el cual, la persona o la familia con riesgo de este trastorno genético, sean informadas de las consecuencias de dicho trastorno y de la probabilidad de tenerlo.
El diagnóstico se basa en la sospecha por los síntomas característicos como la hipotonía, la corta estatura, la hiperfagia, la obesidad, el comportamiento (específicamente con trastornos del tipo obsesivo-compulsivo, el tamaño pequeño de manos y pies, el hipogonadismo y un retraso mental leve. Se confirma con test genéticos, y de hecho se recomienda el uso del test sobre los recién nacidos que presentan una hipotonía significativa.
A nivel molecular, los principales test de Diagnóstico Molecular aplicados en la clínica son:
- Análisis de metilación por PCR: Técnica por la cual si el patrón de metilación hallado corresponde únicamente al materno, se confirma el diagnóstico, que puede ser de PWS asociado a deleciones, disomía uniparental o defectos en impronta.
- Identificación de mutaciones:
- FISH
- PCR cuantitativa
- Estudios de marcadores microsatélites
Con respecto al diagnóstico no molecular, existe un consenso, establecido por primera vez en el año 93 y que fue ratificado en el año 2001
Al tratarse de un cuadro complejo, están implicados diferentes especialidades médicas, Incluyendo genetista, endocrinólogo,neurólogo, neumólogo, oftalmólogo, dermatólogo, traumatólogo y otras especialidades.
OTROS ASPECTOS A TOMAR EN CUENTA
Se incluyen a continuación aspectos relacionados con las capacidades cognitivas, conducta, lenguaje, aprendizaje y escolarización de las personas afectadas..
Aspectos cognitivos
En general, las personas con SPW suelen sufrir alguna limitación cognitiva. Dentro de esta limitación, existen grandes diferencias interindividuales, siendo el cociente intelectual medio alrededor 70. A nivel cognitivo, estas personas normalmente tienen buena memoria a largo plazo, buena organización perceptiva, buena habilidad para reconocer y evaluar relaciones espaciales, buena decodificación y comprensión lectora y un buen vocabulario expresivo. Sin embargo, suelen presentar: falta de procesamiento secuencial de la información, dificultades en la aritmética, pobre memoria a corto plazo (por ejemplo, les cuesta recordar cadenas de información, como pueden ser órdenes por lo que se tacha muchas veces al niño de desobediente), tienen dificultades de atención y concentración y habilidades motoras finas relacionadas con la planificación motriz, el tono o la fuerza.
Conducta
Las personas con SPW suelen tener un patrón conductual específico en el que se observan fluctuaciones significativas según la edad. Los niños pequeños suelen ser alegres, afectuosos, complacientes y cooperadores hasta que sobre los 6 u 8 años se vuelven más rígidos, irritables y emocionalmente más inseguros. Así comienzan a tener conductas como engullir toda la comida disponible, impaciencia, ataques de ira, enfados, distracciones, problemas de comunicación e impulsividad, suelen ser manipuladores, mentirosos, hábiles, caprichosos, egocéntricos; con frecuencia muestran conductas autolesivas y tienen pocas habilidades interpersonales. Las habilidades de cooperación suelen estar más alteradas, aunque éstas mejoran con la edad.
En las actividades de la vida diaria, las personas con SPW se desenvuelven relativamente bien. Destacan especialmente en las habilidades domésticas de preparación de la comida (en los casos en los que se les permite y no les genera demasiado estrés) y en tareas de auto-ayuda.
Lenguaje
El patrón de desarrollo del lenguaje en las personas con Síndrome de Prader Willi es el mismo que el de toda la población aunque el ritmo es más lento. De esta manera, sus primeras palabras suelen aparecer en torno a los dos años y medio y la producción verbal significativa a menudo es escasa antes de los cuatro años.
Algunas de las características más importantes que a nivel lingüístico presentan los sujetos con SPW son distorsiones y simplificaciones de fonemas, tono de voz inadecuado (bajo para las niñas y alto para los niños), vocabulario reducido y dificultades en la generalización de conceptos y en la compresión de oraciones negativas e interrogativas. Por otra parte tienen limitaciones para construir frases ya que esto exige creatividad y la estructuración de la oración suele ser más lenta y las producciones suelen ser incorrectas e incompletas. La falta de comprensión de los mensajes, les lleva muchas veces a mantener conversaciones sin sentido o a la inhibición y al desinterés comunicativos.
El éxito del aprendizaje de la lectura y escritura dependerá de la expresión/comprensión oral alcanzando, así como el grado de afectación motora, en general recuerdan poco de lo leído y tienen dificultades para referirlo en el orden correcto.
Entre los aspectos positivos se incluyen una buena memoria a largo plazo, facilidad para aprender mediante videos e imágenes y habilidad para evaluar relaciones espaciales. Muchos llegan a ser excelentes lectores e incluso leen por placer.
Escolarización
Para los estudiantes con SPW es muy importante la Atención Temprana, ya que tienen unas altas capacidades en el aprendizaje y cuanto antes comience el desarrollo de sus capacidades mediante la intervención educativa, mejor será su desarrollo y calidad de vida futura.
Para la escolarización de estudiante con SPW existen varias alternativas, siendo la más usual y adecuada la escolarización en un centro ordinario, para favorecer así su integración social. Todo ello dependerá las necesidades educativas de cada estudiante así como de los recursos que los centros ordinarios sean capaces de ofrecer para satisfacer dichas necesidades. A la hora de la escolarización se debe tener en cuenta que para que el estudiante desarrolle sus capacidades, el proceso de enseñanza-aprendizaje debe desarrollarse en un ambiente lo más normalizado posible, permitiéndole así la integración en la sociedad.
Todos los profesionales que trabajen con el estudiantes deberán conocer tanto las necesidades educativas que presentan como algunas características médicas de la enfermedad que afectan al rendimiento escolar. Es imprescindible la colaboración de todos los profesionales que atienden al estudiantes, así como el contacto con la familia, que desempeñan un papel principal en la educación de sus hijos.
Algunas características que deben ser tenidas en cuentas por el centro educativo son la presencia de dificultades en el aprendizaje, falta de atención en tareas cotidianas, fuerte necesidad de comida, apnea del sueño que puede ocasionar cansancio o somnolencia en el aula, problemas de conducta, inestabilidad emocional, conductas obsesivas como rascarse o pellizcarse heridas y disfunciones en la temperatura corporal y umbral de dolor alto.
Por ello los profesionales educativos deben estar advertidos de las necesidades que presentan estos alumnos y los compañeros deben ser conscientes de algunos aspectos. Es conveniente seguir ciertas estrategias: no tener comida en clase, consultar periodicamente con la familia, realizar actividades de educación física, trabajar la autoestima e incentivar las relaciones sociales y el compañerismo. Asimismo es recomendable realizar periódicamente revisiones en el dictamen de escolarización ya que las necesidades de estos estudiante varían con el tiempo.
0 comentarios:
Publicar un comentario